Fragment-based drug discovery (FBDD) has become an increasingly vital approach in the rational design of novel therapeutic compounds. This strategy, recognized for its efficiency in identifying starting points for drug development, hinges on the use of small, fragment-like molecules that weakly bind to a target protein. These fragments serve as the foundation for building high-affinity binders, offering significant advantages over traditional methods that begin with larger, drug-like molecules. For researchers involved in programs like the De-foa-0001625 Early Career Research Program, understanding and leveraging cutting-edge techniques in FBDD, such as fragment elaboration, is paramount for advancing drug discovery pipelines.
Compared to conventional methodologies, FBDD offers enhanced control over the physicochemical properties of resulting molecules right from the outset. Starting with low molecular weight fragments allows for finer adjustments in molecule design, leading to more optimized drug candidates. Furthermore, studies have demonstrated the remarkable efficiency of fragment libraries in chemical space exploration, achieving hit rates significantly higher—10 to 1000 times greater—than traditional high-throughput screening assays. This efficiency translates to a higher probability of identifying a viable starting point and better management of the subsequent optimization phases in drug development, crucial aspects for projects supported by the DE-FOA-0001625 initiative which often emphasizes innovative and efficient research methodologies.
Following the identification of initial fragment hits, lead molecule development typically involves three primary strategies: elaboration (or growing), fragment linking, and fragment merging. Elaboration, the focus of this discussion, involves extending a single fragment by adding functional groups to create more favorable interactions with the target protein. Fragment linking connects two concurrently bound fragments with a molecular bridge, effectively combining them into a single molecule. Fragment merging designs molecules incorporating motifs from two or more fragments that bind in overlapping regions.
Currently, the design process for these strategies largely relies on the expertise of medicinal chemists, who use computational tools and their chemical intuition to propose promising molecular designs. However, this human-centric approach can be limited by cognitive biases and the sheer volume of data from large fragment screens, making it challenging to objectively assess all potential elaboration opportunities. This is where innovative computational methods become invaluable, particularly for early career researchers in programs like DE-FOA-0001625 who are encouraged to explore and develop novel approaches to accelerate drug discovery.
The field has witnessed a surge of interest in machine learning models aimed at rapidly generating and screening vast libraries of molecules for drug candidacy. These models employ various molecular representations, including SMILES, graphs, and atomic density grids, alongside advanced deep learning architectures like generative adversarial networks and variational autoencoders. To optimize molecular properties, techniques for multiobjective optimization, such as reinforcement learning and Bayesian optimization, are also being explored.
While early generative models focused on de novo molecule generation, recent efforts have shifted towards enhancing the efficiency of fragment-to-lead campaigns using deep learning. Graph-based approaches and SMILES-based models like Scaffold-Decorator have been developed to elaborate fragments. These methods, while innovative, often lack the ability to consider protein structural context or user-defined design parameters, which are crucial for ensuring elaborations are structurally relevant and fit within the protein binding pocket. DEVELOP, a previous model, introduced pharmacophoric constraints and length control, offering improved control but still requiring pre-existing knowledge of active molecules or human-specified constraints. Similarly, LibINVENT focuses on core-sharing chemical libraries and 3D similarity to known actives. These limitations highlight the need for methods that can directly leverage protein structure information for more targeted and efficient fragment elaboration – a gap that the DE-FOA-0001625 Early Career Research Program encourages researchers to address.
In contrast to generative approaches, database-driven methods like CReM and structure-aware methods like FragRep and DeepFrag have emerged. CReM utilizes a database of molecular fragments to identify potential elaborations based on local chemical context. FragRep and DeepFrag incorporate protein-specific information to refine ligand modifications. For de novo molecule generation directly informed by protein structure, models like those by Skalic et al. and Masuda et al. use GANs and latent representations to generate ligands complementary to the binding pocket. Kim et al. employed water pharmacophore models to guide molecule generation. However, these models often lack the flexibility to integrate user-defined design hypotheses or require target-specific training datasets, limiting their broad applicability in diverse drug discovery settings often encountered in research funded by initiatives like DE-FOA-0001625 Early Career Research Program.
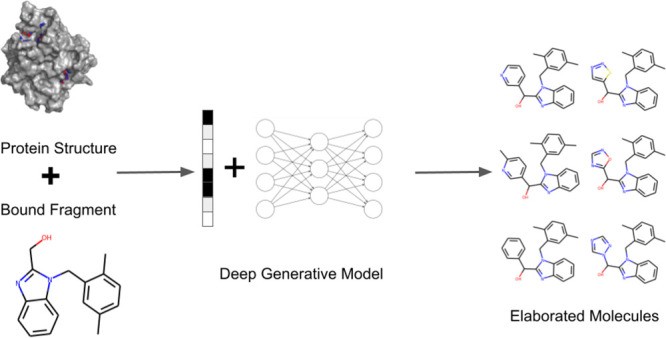
This article introduces STRIFE (Structure Informed Fragment Elaboration), a novel generative model specifically designed for fragment elaboration. STRIFE distinguishes itself by extracting interpretable structural information directly from the protein target and using this information to guide the generation of elaborations. Unlike existing fragment-based generative models that either implicitly learn from known ligands or ignore protein structure, STRIFE directly leverages protein-derived insights. Furthermore, STRIFE offers user customizability, allowing researchers to incorporate their design hypotheses alongside protein-derived information. Through rigorous evaluation, STRIFE demonstrates significant improvements over existing fragment elaboration methods in generating ligand-efficient elaborations. Its applicability to real-world FBDD campaigns is further validated through case studies, including the elaboration of a fragment bound to N-myristoyltransferase and the exploration of side-chain flexibility in tumor necrosis factor inhibition. This advancement in fragment elaboration technology holds significant promise for researchers in the DE-FOA-0001625 Early Career Research Program and the broader drug discovery community, offering a powerful tool to accelerate the development of novel therapeutics.
